User’s Guide#
The skeleton_keys package supports the skeletal analysis of neuron morphologies.
It is used to perform a series of analysis steps in a consistent and scaleable
manner. It can be used to align a series of morphologies from the isocortex to a
set of reference layer depths/thicknesses, create layer-aligned depth profile
histograms, and calculate morphological features using the neuron_morphology package.
The main inputs for skeleton_keys are neuron morphologies in the form of
SWC files, layer drawings for the cells of interest, and reference information
such as a reference set of layer depths/thicknesses. The package contains
several command line scripts that are typically used in sequence to process a
set of morphologies and end up with a comparable set of morphological features
for the entire set.
Aligning a morphology to a reference set of layers#
The depths and thicknesses of cortical layers can vary from location to location across the isocortex. However, some cells send processes into specific layers regardless of their location. Analyses that use the cells only at their original size may blur precise distinctions that follow layer boundaries; therefore, several features calculated by this package rely on first aligning morphologies to a consistent set of layer thicknesses/depths. These layer-aligned morphologies can also be used for other purposes, like visualization.
Since the isocortex is a curved structure, the depth is not always straightforward
to calculate. The neuron_morphology package (used by skeleton_keys to
perform the layer alignemnt) takes the approach used in the
Allen Common Coordinate Framework for the mouse brain,
where paths traveling through isopotential surfaces between the pia and white matter
define the depth dimension of the cortex.
Therefore, to align to a common set of layers, we need information about
where the pia, white matter, and layers are with respect to the cell of interest.
At the Allen Institute for Brain Science, these are drawn on 20x images
of the morphology, using a DAPI stain to identify layers. These lines and polygons
become inputs into the skeleton_keys scripts.
These layer drawings match the orientation of the reconstructed cell taken directly from the image (i.e., before any rotation to place pia upward is done).
As an example, we will use an Sst+ inhibitory neuron from the Gouwens et al. (2020) study
where we have also saved the layer drawings in the example_data directory as
a JSON file in the format expected by skeleton_keys.
In its original orientation, the morphology looks like this:
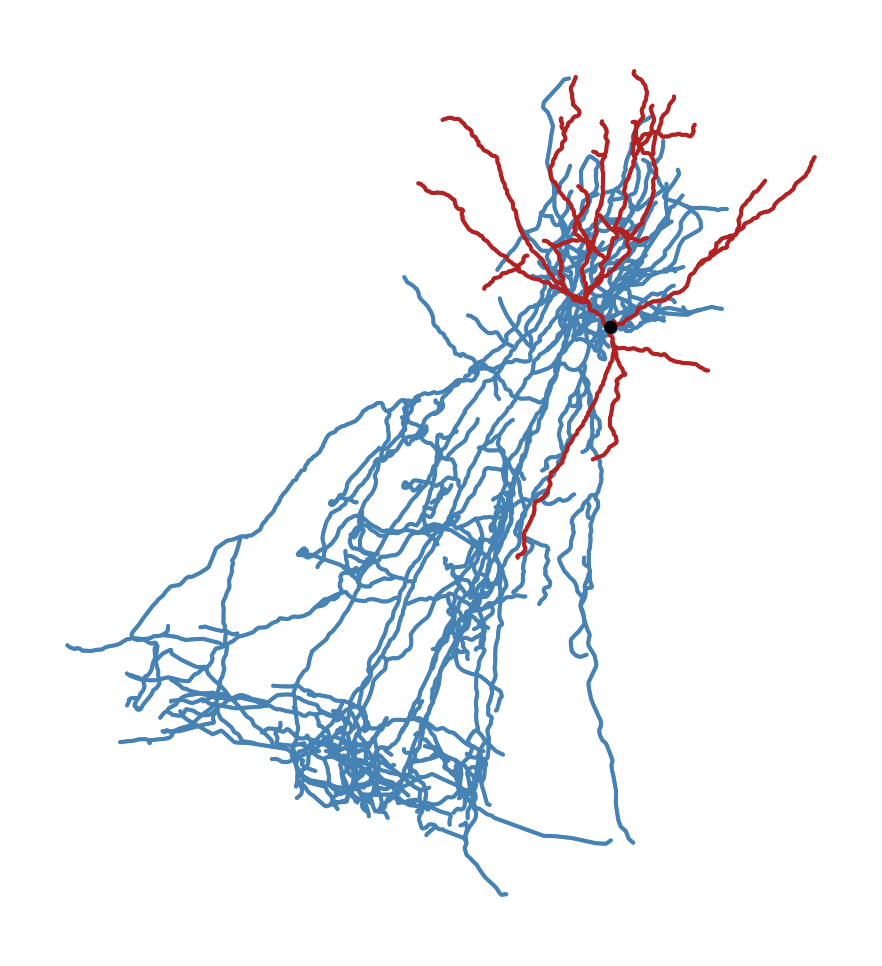
Sst neuron in original orientation. Dendrite is red, axon is blue, soma is black dot.#
Our layer drawings (with a matched orientation) look like:
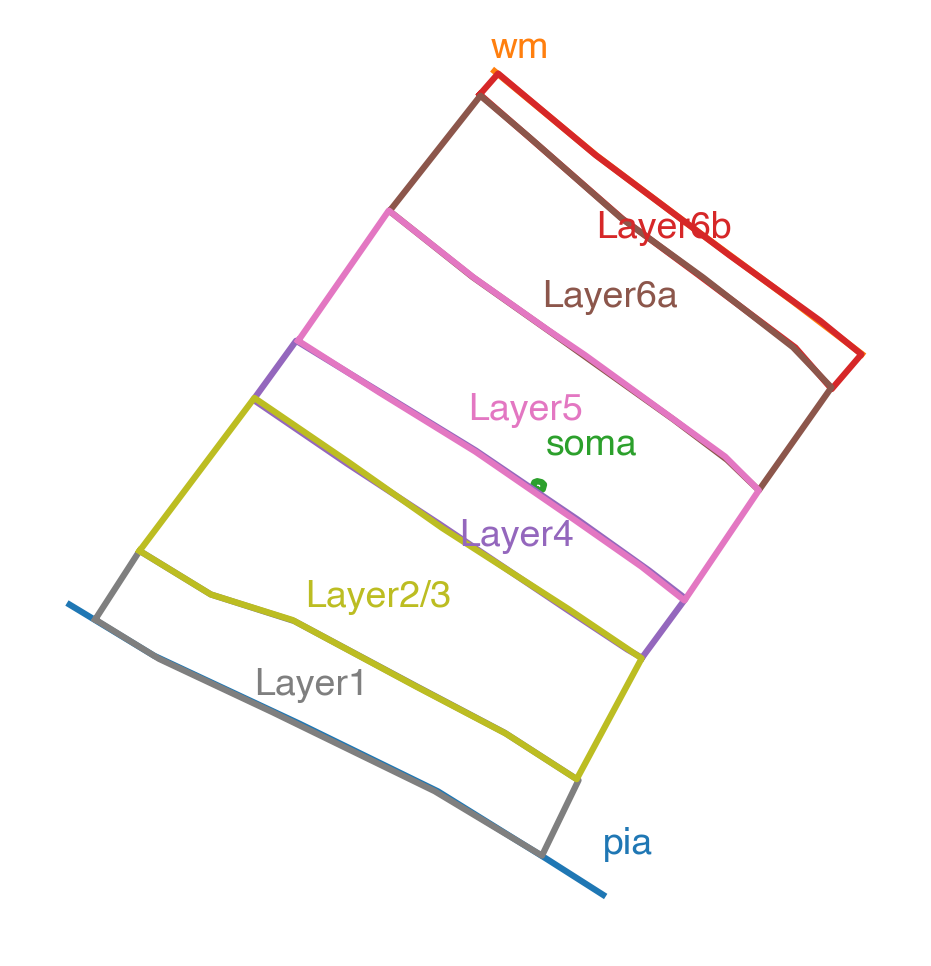
Pia, white matter, soma outline, and layer drawings#
And together (after aligning the soma locations) they look like:
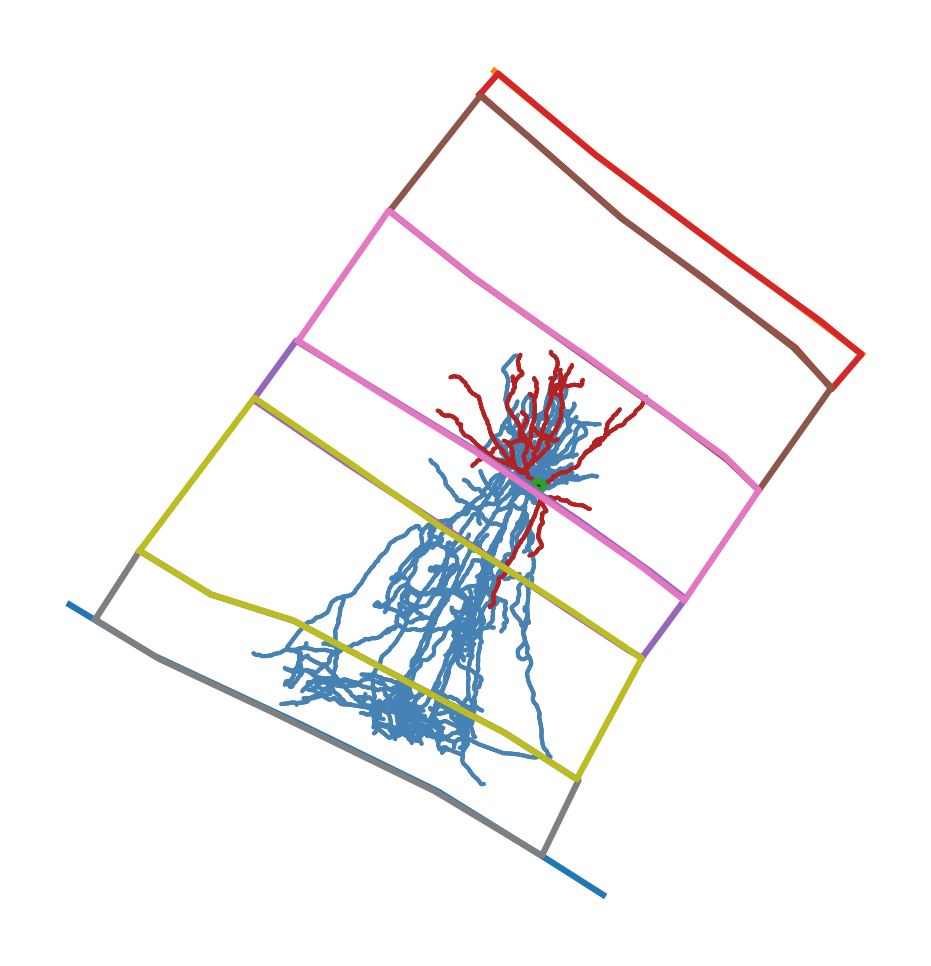
Morphology and layer drawings in original orientation#
Note: This script also has functionality to correct the morphology for shrinkage (which can happen because the fixed tissue that is image dries out and becomes flatter than the original) and slice angle tilt (which happens when the cutting angle for brain slices does not match the curvature of that part of the brain). However, these features are currently written to require access to an internal Allen Institute database and do not yet have alternative input formats. Therefore, we will not use those functions in this guide.
We will supply the script with the following inputs:
- specimen_id - an integer identifier for the cell.
Here, it is primarily used to access internal database information (which we aren’t doing), but in other scripts it is used to associate this cell with its features. Here, the specimen ID of our example is
740135032.
- swc_path - a file path to the SWC file in its original orientation.
Here, our example cell’s SWC file is
Sst-IRES-Cre_Ai14-408415.03.02.02_864876723_m.swc.
- surface_and_layers_file - a JSON file with the layer drawings.
Here, our example uses the file
740135032_surfaces_and_layers.json.
- layer_depths_file - a JSON file with the set of layer depths we’re aligning the cell to.
In this case we’ll use an average set of depths included as a test file,
avg_layer_depths.json.
Therefore, our command will be:
skelekeys-layer-aligned-swc --specimen_id 740135032 \
--swc_path Sst-IRES-Cre_Ai14-408415.03.02.02_864876723_m.swc \
--surface_and_layers_file 740135032_surfaces_and_layers.json \
--layer_depths_file avg_layer_depths.json \
--correct_for_shrinkage False \
--correct_for_slice_angle False \
--output_file layer_aligned_740135032.swc
This creates a new SWC file (layer_aligned_740135032.swc) that is (1)
uprighted and (2) stretched or squished to align each of its points to the
reference set of layers.
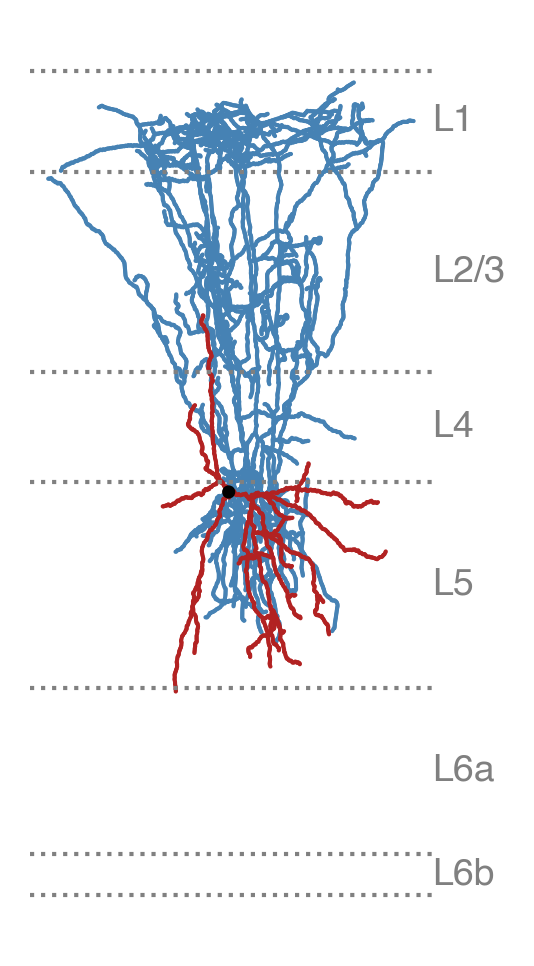
Layer-aligned morphology#
Uprighting a morphology without layer alignment#
If you only want to orient morphology so that pia is up and the white matter is
down, but without making any layer thickness adjustments, you can use the
command-line utility skelekeys-upright-corrected-swc. It still requires
layer drawings, though, to know which direction the pia and white matter are
relative to the originally reconstructed morphology.
Note: This script, too, can correct the morphology for shrinkage and slice angle tilt, but we are again skipping that since it currently can only use internally databased information.
It takes a similar set of arguments as before (but notably without the --layer_depths_file
argument).
skelekeys-upright-corrected-swc --specimen_id 740135032 \
--swc_path Sst-IRES-Cre_Ai14-408415.03.02.02_864876723_m.swc \
--surface_and_layers_file 740135032_surfaces_and_layers.json \
--correct_for_shrinkage False \
--correct_for_slice_angle False \
--output_file upright_only_740135032.swc
The output looks like:
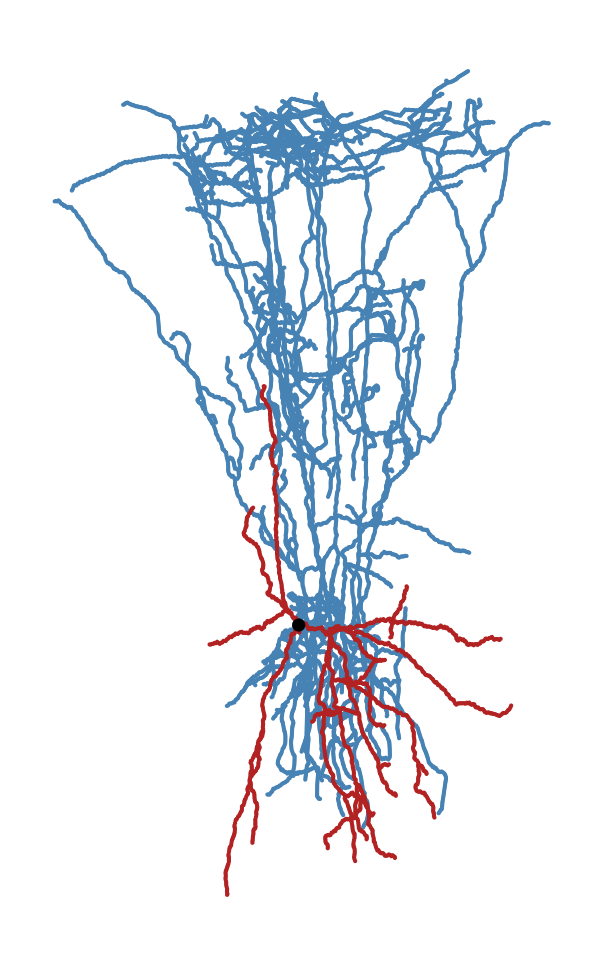
Upright (but not layer-aligned) morphology#
Calculating depth profiles#
A relevant aspect for distinguishing morphologies is the depth profile, which is a 1D histogram of the number of nodes across a set of depth bins, divided by compartment type (i.e., axon, basal dendrite, apical dendrite). These can be used to calculate reduced-dimesion representations of those profiles, determine the overlap of different compartment types, etc.
The command line utility skelekeys-profiles-from-swcs will create a CSV file of
depth profiles from a list of layer-aligned SWC files.
The script expects the layer-aligned SWC files to be in a single directory (--swc_dir)
and be named as {specimen_id}.swc. For this example, we have moved the layer-aligned
Sst cell’s SWC file (740135032) into another directory and renamed it; we have also layer aligned
a Pvalb cell (606271263) and a Vip cell (694146546).

Layer-aligned Sst cell, Pvalb cell, and Vip cell#
To run the script for our example, we give it the following inputs:
- specimen_id_file - a text file with specimen IDs
The text file has one integer ID per line. Here we’re using
example_specimen_ids.txt.
- swc_dir - the directory of layer-aligned SWC files
Here our directory is
layer_aligned_swcs
- layer_depths_file - a JSON file with the reference set of layer depths
This is so that the script knows where the white matter begins (so that it can determine how far past to include)
- output_hist_file - an output CSV file path
This CSV file contains the depth histograms - the columns are depth bins, and the rows are cells
- output_soma_file - an output CSV file path
The script also saves the layer-aligned soma depths, which is used by other command line scripts in the package
Our command will then be:
skelekeys-profiles-from-swcs --specimen_id_file example_specimen_ids.txt \
--swc_dir layer_aligned_swcs \
--layer_depths_file avg_layer_depths.json \
--output_hist_file aligned_depth_profiles.csv \
--output_soma_file aligned_soma_depths.csv
This produces the following depth profiles:
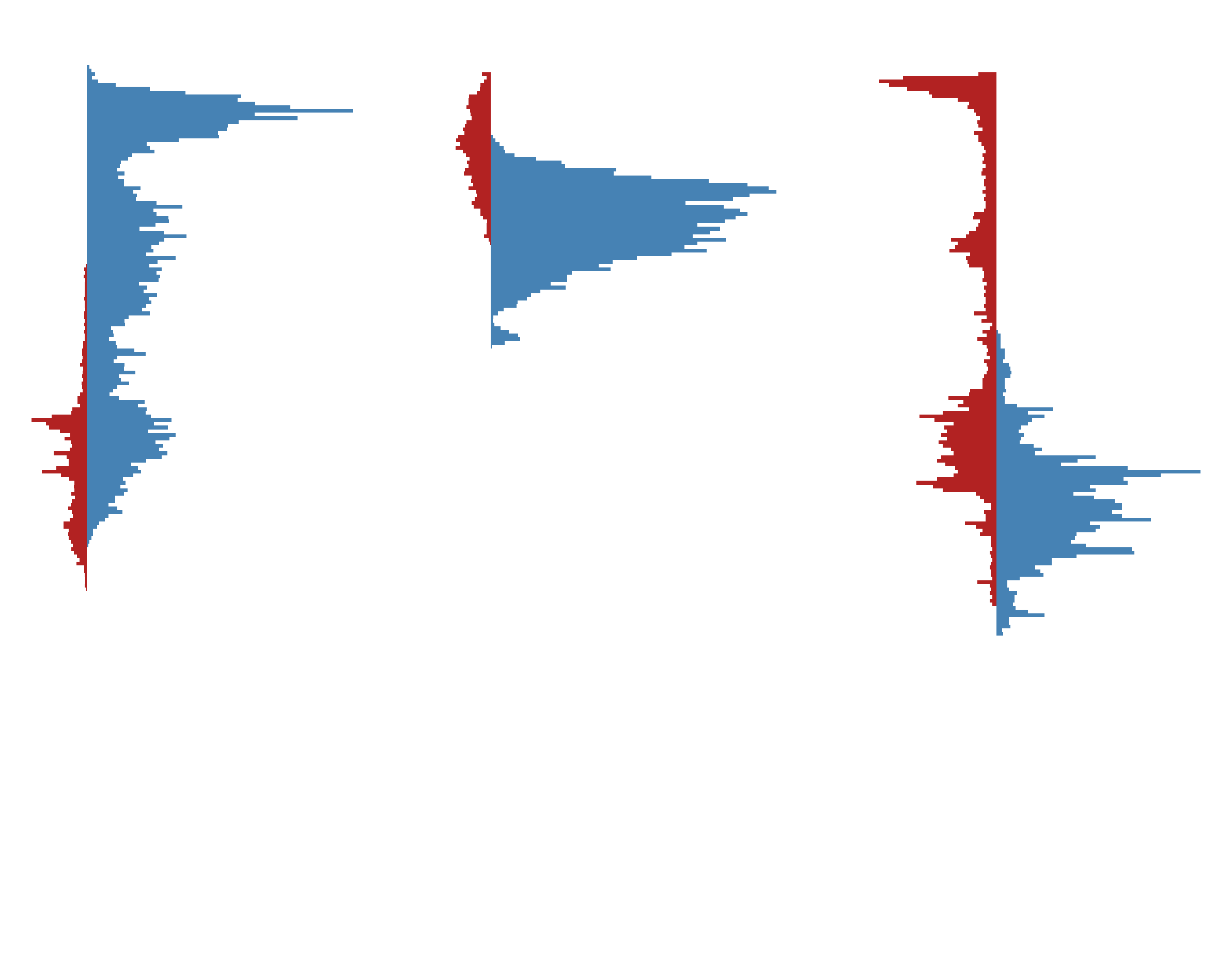
Layer-aligned depth profiles of the three example cells#
PCA on depth profiles#
A reduced dimension representation of the depth profiles can serve as useful
features for distinguishing morphologies. However, analyses like PCA will produce
loadings that vary depending on the data set. The command line utility
skelekeys-calc-histogram-loadings can be used to generate a fixed loading file
from a set of morphologies that can be used to analyze other morphologies with
other scripts.
If you simply want to use the PCA loadings for a new set of cells and not
transfer those loadings to another set, you do not need to use this utility
(you can use the skelekeys-morph-features script by itself). That script
will also let you save the loadings. But this script will allow you to calculate
loadings without having to calculate all the other morphological features.
In this example, we will calculate and save the PCA loadings for the axonal compartments. The command to do so is:
skelekeys-calc-histogram-loadings --specimen_id_file example_specimen_ids.txt \
--aligned_depth_profile_file aligned_depth_profiles.csv \
--analyze_axon True \
--save_axon_depth_profile_loadings_file axon_loadings.csv
Calculating morphological features#
Once we have the layer depths files (and optionally a set of pre-calculated loadings),
we can calculate a set of morphological features for each cell using the
command line utility skelekeys-morph-features.
The main inputs are the set of upright (but not layer-aligned) SWC files and the depth
profile CSV file. We also specify which compartments we want to analyze (for example
if we have a set of excitatory neurons that don’t have reconstructed local axons,
we would not want to analyze axonal compartments).
To continue our example, we will analyze the features using the following command:
skelekeys-morph-features --specimen_id_file example_specimen_ids.txt \
--swc_dir upright_swcs \
--aligned_depth_profile_file aligned_depth_profiles.csv \
--aligned_soma_file aligned_soma_depths.csv \
--analyze_axon True \
--analyze_basal_dendrite True \
--analyze_apical_dendrite False \
--output_file example_features_long.csv
This produces a long-form feature data file.
specimen_id |
feature |
compartment_type |
dimension |
value |
|
|---|---|---|---|---|---|
0 |
740135032 |
aligned_dist_from_pia |
soma |
none |
488.95489922056464 |
1 |
606271263 |
aligned_dist_from_pia |
soma |
none |
185.35510042590795 |
2 |
694146546 |
aligned_dist_from_pia |
soma |
none |
363.1829539564272 |
3 |
740135032 |
depth_pc_0 |
axon |
none |
-591.3768052788041 |
4 |
740135032 |
depth_pc_1 |
axon |
none |
771.0611928161894 |
5 |
606271263 |
depth_pc_0 |
axon |
none |
1557.1882340570776 |
6 |
606271263 |
depth_pc_1 |
axon |
none |
-114.4320436027042 |
7 |
694146546 |
depth_pc_0 |
axon |
none |
-965.8114287782732 |
8 |
694146546 |
depth_pc_1 |
axon |
none |
-656.6291492134859 |
9 |
740135032 |
emd_with_basal_dendrite |
axon |
none |
53.842009790570955 |
10 |
606271263 |
emd_with_basal_dendrite |
axon |
none |
20.02095840833445 |
11 |
694146546 |
emd_with_basal_dendrite |
axon |
none |
50.38248981181829 |
12 |
740135032 |
frac_above_basal_dendrite |
axon |
none |
0.5537837260625045 |
13 |
740135032 |
frac_intersect_basal_dendrite |
axon |
none |
0.4462162739374956 |
14 |
740135032 |
frac_below_basal_dendrite |
axon |
none |
0.0 |
15 |
606271263 |
frac_above_basal_dendrite |
axon |
none |
0.0 |
16 |
606271263 |
frac_intersect_basal_dendrite |
axon |
none |
0.7648047321368555 |
17 |
606271263 |
frac_below_basal_dendrite |
axon |
none |
0.23519526786314446 |
18 |
694146546 |
frac_above_basal_dendrite |
axon |
none |
0.0 |
19 |
694146546 |
frac_intersect_basal_dendrite |
axon |
none |
0.9692556634304207 |
20 |
694146546 |
frac_below_basal_dendrite |
axon |
none |
0.030744336569579287 |
21 |
740135032 |
frac_above_axon |
basal_dendrite |
none |
0.0 |
22 |
740135032 |
frac_intersect_axon |
basal_dendrite |
none |
0.946916890080429 |
23 |
740135032 |
frac_below_axon |
basal_dendrite |
none |
0.05308310991957105 |
24 |
606271263 |
frac_above_axon |
basal_dendrite |
none |
0.37383177570093457 |
25 |
606271263 |
frac_intersect_axon |
basal_dendrite |
none |
0.6261682242990654 |
26 |
606271263 |
frac_below_axon |
basal_dendrite |
none |
0.0 |
27 |
694146546 |
frac_above_axon |
basal_dendrite |
none |
0.47685185185185186 |
28 |
694146546 |
frac_intersect_axon |
basal_dendrite |
none |
0.5231481481481481 |
29 |
694146546 |
frac_below_axon |
basal_dendrite |
none |
0.0 |
30 |
740135032 |
extent |
basal_dendrite |
x |
259.0984945349686 |
31 |
740135032 |
extent |
basal_dendrite |
y |
360.15791867251323 |
32 |
740135032 |
bias |
basal_dendrite |
x |
105.9123765215528 |
33 |
740135032 |
bias |
basal_dendrite |
y |
-5.017693772805842 |
Post-processing morphological features#
The long-form file can be used for many purposes, but it is also useful to convert the data to a wide format where the features are the columns and the rows are cells. At the same time, it can also be useful to normalize the different features for analyzes like classification and clustering.
The command line utility skelekeys-postprocess-features is used to perform
these operations.
skelekeys-postprocess-features \
--input_files "['example_features_long.csv']" \
--wide_normalized_output_file example_features_wide_normalized.csv \
--wide_unnormalized_output_file example_features_wide_unnormalized.csv
Note the syntax for passing a list of files for the argschema
command line argument. If you passed more than one file (for example, to
normalize features calculated from two sets of cells to the same scale), you
would separate each argument with a comma (as in "['file_one.csv','file_two.csv']").
The wide form feature file output looks like:
specimen_id |
axon_bias_x |
axon_bias_y |
axon_depth_pc_0 |
axon_depth_pc_1 |
axon_emd_with_basal_dendrite |
axon_exit_distance |
axon_exit_theta |
axon_extent_x |
axon_extent_y |
axon_frac_above_basal_dendrite |
axon_frac_below_basal_dendrite |
axon_frac_intersect_basal_dendrite |
axon_max_branch_order |
axon_max_euclidean_distance |
axon_max_path_distance |
axon_mean_contraction |
axon_num_branches |
axon_num_outer_bifurcations |
axon_soma_percentile_x |
axon_soma_percentile_y |
axon_total_length |
basal_dendrite_bias_x |
basal_dendrite_bias_y |
basal_dendrite_calculate_number_of_stems |
basal_dendrite_extent_x |
basal_dendrite_extent_y |
basal_dendrite_frac_above_axon |
basal_dendrite_frac_below_axon |
basal_dendrite_frac_intersect_axon |
basal_dendrite_max_branch_order |
basal_dendrite_max_euclidean_distance |
basal_dendrite_max_path_distance |
basal_dendrite_mean_contraction |
basal_dendrite_mean_diameter |
basal_dendrite_num_branches |
basal_dendrite_num_outer_bifurcations |
basal_dendrite_soma_percentile_x |
basal_dendrite_soma_percentile_y |
basal_dendrite_stem_exit_down |
basal_dendrite_stem_exit_side |
basal_dendrite_stem_exit_up |
basal_dendrite_total_length |
basal_dendrite_total_surface_area |
none_early_branch_path |
soma_aligned_dist_from_pia |
soma_surface_area |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
606271263 |
56.22872989248452 |
-121.2103061622121 |
1557.1882340570776 |
-114.4320436027042 |
20.02095840833445 |
0.0 |
0.6246170133542251 |
400.4118094004192 |
262.2553175261422 |
0.0 |
0.2351952678631444 |
0.7648047321368555 |
25.0 |
240.6846227360817 |
902.0221232091972 |
0.8194136640160532 |
665.0 |
1.845098040014257 |
0.4889426631713383 |
0.7447065940713854 |
16935.227964908227 |
0.1618829328459696 |
107.3874873605935 |
7.0 |
292.8979665049092 |
227.9213594244781 |
0.3738317757009345 |
0.0 |
0.6261682242990654 |
5.0 |
221.48224685084813 |
261.57785404864563 |
0.8777370205959968 |
0.5797508521165475 |
35.0 |
0.3010299956639812 |
0.4870808136338647 |
0.1423859263331501 |
0.4285714285714285 |
0.5714285714285714 |
0.0 |
2057.344684556989 |
3746.221348808403 |
0.5122275147209207 |
185.35510042590795 |
334.48341751636667 |
694146546 |
20.979837762279885 |
-337.62296125250555 |
-965.8114287782732 |
-656.6291492134859 |
50.38248981181829 |
8.953753130391712 |
0.8624144236833117 |
456.4507565320748 |
329.4824306809105 |
0.0 |
0.0307443365695792 |
0.9692556634304208 |
14.0 |
369.0094227941884 |
537.3808743109979 |
0.8804317016691116 |
117.0 |
1.462397997898956 |
0.3268608414239482 |
1.0 |
3856.927925804772 |
4.519987715724085 |
45.38310515181024 |
3.0 |
158.97060160570243 |
640.1652005614803 |
0.4768518518518518 |
0.0 |
0.5231481481481481 |
9.0 |
353.0402965262322 |
434.5470290069107 |
0.896216086452986 |
0.6016030092592592 |
69.0 |
1.1760912590556813 |
0.1689814814814815 |
0.5262345679012346 |
0.3333333333333333 |
0.0 |
0.6666666666666666 |
3185.736311407383 |
5983.0167307564 |
0.7175151344782787 |
363.1829539564272 |
240.61616017724623 |
740135032 |
5.169444355631811 |
297.6111166818944 |
-591.3768052788041 |
771.0611928161894 |
53.84200979057096 |
20.438908268544992 |
0.5210092861882935 |
424.4629900629744 |
587.2419117540196 |
0.5537837260625045 |
0.0 |
0.4462162739374956 |
24.0 |
465.7975422331148 |
712.7996048944084 |
0.8629205591762148 |
475.0 |
2.05307844348342 |
0.3885527158823948 |
0.1986189221897914 |
17053.86846302751 |
105.9123765215528 |
-5.017693772805842 |
4.0 |
259.0984945349686 |
360.15791867251323 |
0.0 |
0.053083109919571 |
0.946916890080429 |
9.0 |
196.6200075357029 |
228.6181419435221 |
0.8927636513643621 |
0.4823025201072386 |
44.0 |
0.7781512503836436 |
0.3179624664879356 |
0.8273458445040215 |
0.0 |
1.0 |
0.0 |
2204.244429816128 |
3338.8595673807986 |
0.9062144295279364 |
488.95489922056464 |
201.46425475133609 |
Working with general coordinates#
The skeleton_keys package was originally built around processing SWC files,
but it can also be used to align arbitrary sets of coordinates, if provided with
the appropriate layer drawings.
The command line utility skelekeys-layer-aligned-coords can take a CSV and
adjust the specified coordinates to make them layer-aligned. It takes the following
inputs:
- coordinate_file - a CSV with coordinates
The coordinate columns must contain “x”, “y”, and “z” in their names, but can have prefixes and/or suffixes (see below). We’ll use an example file named
coord_example.csv.
- layer_depths_file a JSON file with the set of layer depths we’re aligning the cell to.
Here again we’ll use an average set of depths included as a test file,
avg_layer_depths.json.
- surface_and_layers_file - a JSON file with the layer drawings.
Here, our example uses the file
coord_layer_drawings.json.
- coordinate_column_prefix (and/or _suffix) - strings of common prefixes/suffixes
This allows the coordinate columns to have names other than the default
x,y, andz, but in this example we do not need to use them.
Using this, we can take a starting example file (the columns cell_id and
target_cell_type contain extra metadata about the coordinates):
cell_id |
target_cell_type |
x |
y |
z |
|---|---|---|---|---|
1 |
4P |
838.184 |
691.792 |
821.64 |
1 |
4P |
850.392 |
676.904 |
796.56 |
2 |
4P |
741.704 |
649.608 |
737.76 |
3 |
5P-ET |
739.656 |
680.464 |
950.92 |
3 |
5P-IT |
762.28 |
748.584 |
826.44 |
Use the command:
skelekeys-layer-aligned-coords \
--coordinate_file coord_example.csv \
--surface_and_layers_file coord_layer_drawings.json \
--layer_depths_file avg_layer_depths.json \
--output_file aligned_coord_example.csv
And obtain:
cell_id |
target_cell_type |
x |
y |
z |
|---|---|---|---|---|
1 |
4P |
-92.0060469028378 |
-421.0797644340709 |
821.64 |
1 |
4P |
-105.11025186647323 |
-399.30901042554444 |
796.56 |
2 |
4P |
1.6844004295792985 |
-335.2109182277234 |
737.76 |
3 |
5P-ET |
5.634215690400225 |
-382.48922026392205 |
950.92 |
3 |
5P-IT |
-12.739372718728555 |
-508.88802416730545 |
826.44 |
Note that the x values have also changed because we have rotated the
coordinates to an upright orientation (with pia at the top).
This aligned coordinate file can be used to generate histograms with the
skelekeys-profiles-from-coords command line utility. You need to specify
which column contains the depth values (here, the column labeled y) with the
depth_label argument.
You can use other columns in the CSV file to split the histograms across rows using the
--index_label argument, and/or you can create multiple histograms per row (as in the
compartment type histograms above) with the --hist_split_label argument.
For example:
skelekeys-profiles-from-coords \
--coordinate_file aligned_coord_example.csv \
--layer_depths_file avg_layer_depths.json \
--depth_label y \
--index_label cell_id \
--hist_split_label target_cell_type \
--output_hist_file aligned_coord_hist.csv